Dautres protéinopathies sont identifiées : entre autres exemples ; lHuntingtine dans la Chorée de Huntington, maladie familiale de transmission autosomique dominante, appartenant au groupe des Maladies par Expansion de Polyglutamine dont on connaît une trentaine de formes parmi lesquelles lAtrophie Musculaire Bulbaire et Spinale, lAtrophie Dentato-Rubro-Pallido-Luysienne, les Ataxies Spino Cérebelleuses SCA 1,2,3,6,7,17. Le gène codant pour la Huntingtine est situé sur le chromosome 4 (1993), la mutation consiste en une expansion du codon GAG.

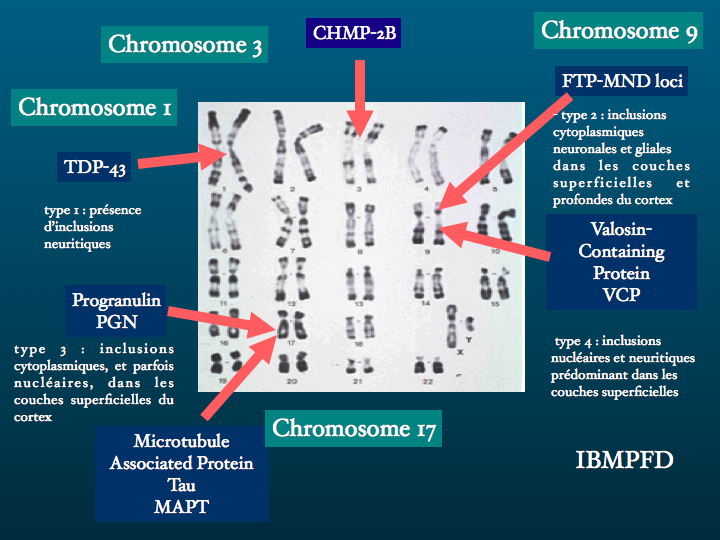
LHérédo-Ataxie de Friedreich caractérisée par la présence de Frataxine est une maladie de transmission autosomale récessive dont le gène est situé sur le Chromosome 9 (1988), et qui se caractérise par la présence de Frataxine (1996). La mutation consiste en une expansion dun nucléotide GAA dans lintron 1 du gène codant pour la frataxine FRDA. Le développement chez ces patients dune Cardiomyopathie, dun diabète illustre le fait quune pathologie neurodégénérative concerne également dautres tissus comme ici les cellules myocardiques ou les cellules pancréatiques.

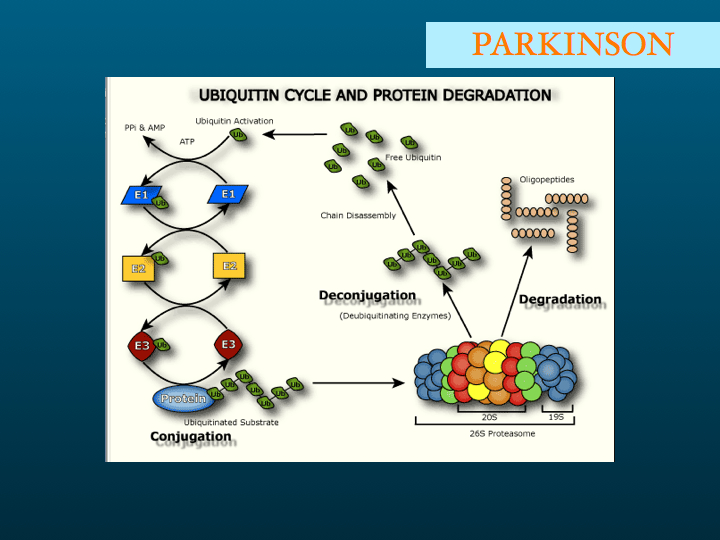
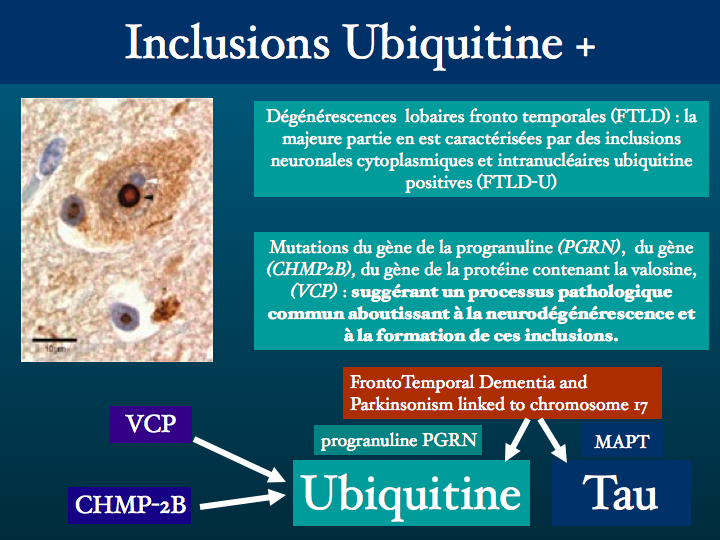
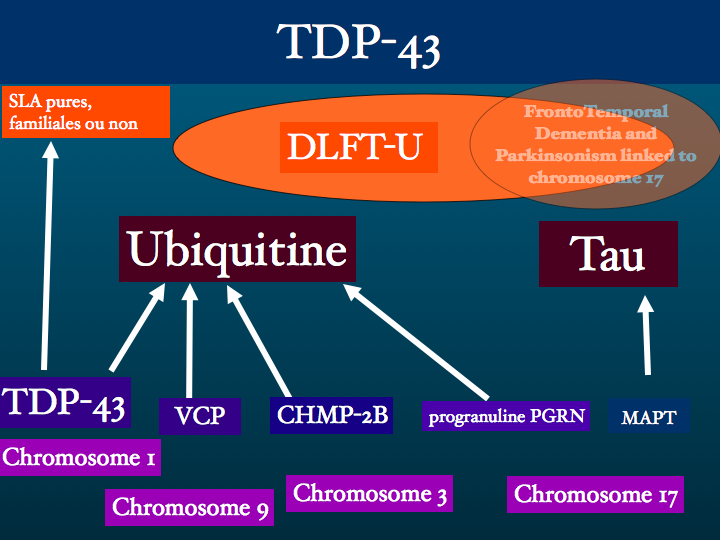
Nous développerons par la suite la question des inclusions ubiquitine +. Pour l'instant nous proposons d'aller plus loin dans le chapitre des maladies avec inclusions Tau +, les tauopathies.


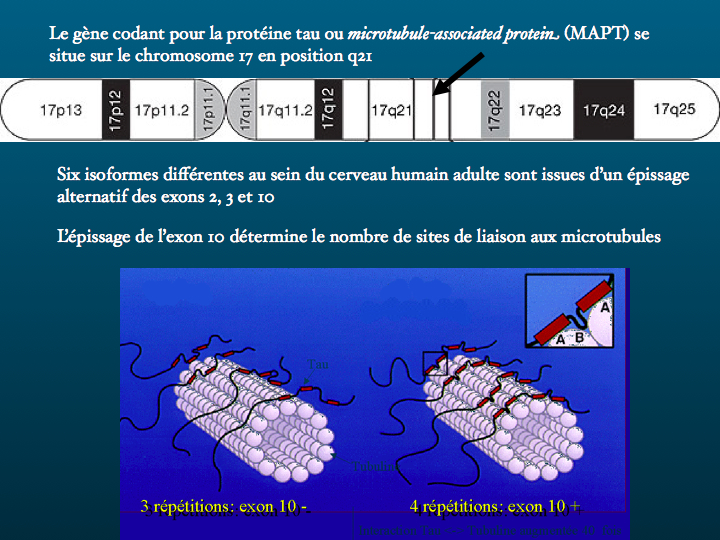
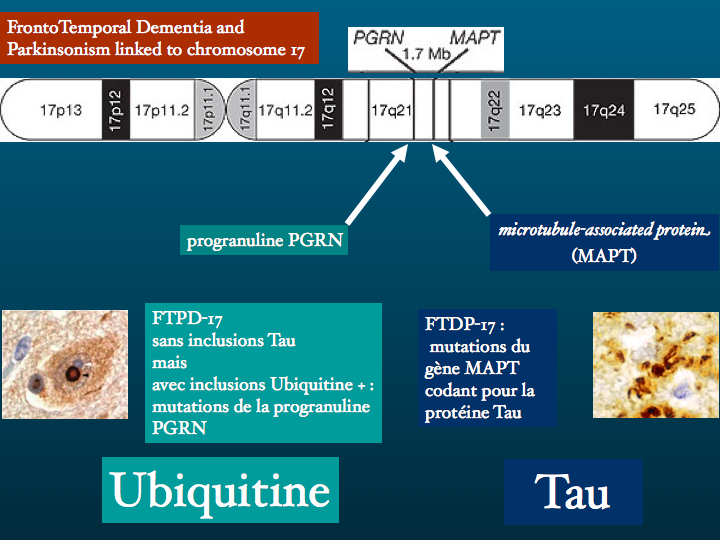
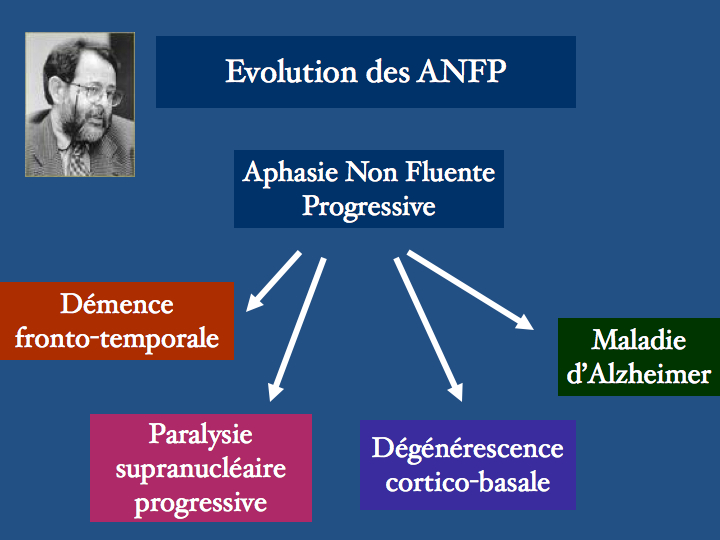
La protéine Tau ou Microtubule-associated protein ( MAPT) est codée par un gène situé sur le chromosome 17. Il existe en fait plusieurs protéines Tau isoformes, issues d'un processus d'épissage alternatif. Leur fonction la mieux connue est la stabilisation des microtubule consitutives du cytosquelette. Ces microtubules sont des structures labiles, les protéines Tau se fixent ainsi que le schéma le représente, par trois ou par quatre points, selon l'isoforme qui les caractérise. Conséquences d'un défaut de production ou d'une accumulation pathologique, les tauopathies sont nombreuses, les plus fréquentes étant la maladie d'Alzheimer, certaines démences fronto-temporales, la paralysie supranucléaire progressive, la dégénérescence cortico-basale.
Nous supposons que la synthèse des protéines n'a pas de secret pour vous, néanmoins il est peut-être bon de préciser au préalable ce qu'est un épissage alternatif, sinon ce qui suit risque de paraître obscur.
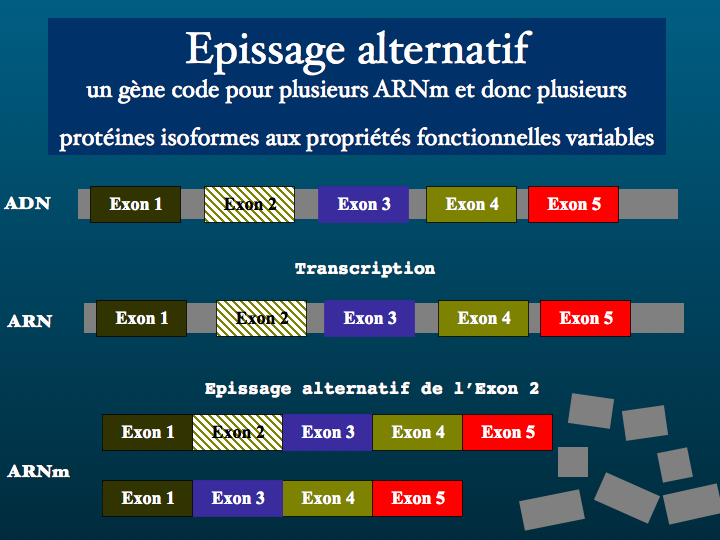
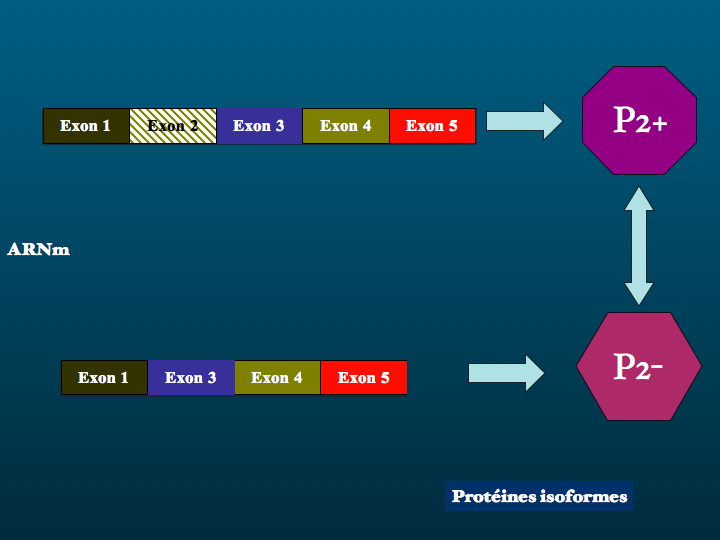
Les gènes codant des protéines sont constitués d'une suite alternée d'exons et d'introns. L'épissage est un processus qui se déroule dans les spliceosomes ( analogues des ribosomes cytoplasmiques ) du noyau cellulaire et consiste à exclure les introns et lier les exons. L'expression ou non d'un ou plusieurs exons lors de la transcription de l'ARN messager détermine la structure de la protéine produite en fin de processus. Et donc sa fonction. Un point important à préciser dès maintenant est que la coexistence d'isoformes est un équilibre que l'augmentation ou la diminution d'un seul parmi ces isoformes suffit à menacer.
Le gène Dscam de la drosophile peut coder jusqu'à 38016 ARN messagers différents.
Prenons maintenant l'exemple d'un exon 2 exprimé ou non exprimé lors de l'épissage alternatif, qui correspond en fait à la situation rencontrée ici lors de l'épissage alternatif de la protéine tau, pour les exons 2,3 et 10 :
1. Les microtubules sont des filaments du cytosquelette, nécessaires au transport des matériaux synthétisés par le corps cellulaire vers les terminaisons nerveuses.
2. Les microtubules sont constitués par l'assemblage de dimères de tubuline.
3. Les microtubules sont des structures labiles, stabilisées par les protéines tau.
4. Le gène Tau est un long gène de 100 kb situé sur le chromosome 17 :
A: six différents mRNAs sont produits dans le cerveau adulte, par épissage alternatif
B: en conséquence six isoformes de Tau sont exprimés, dont trois possèdent un site de lien additionnel aux microtubules, correspondant à la séquence encodée par lexon 10
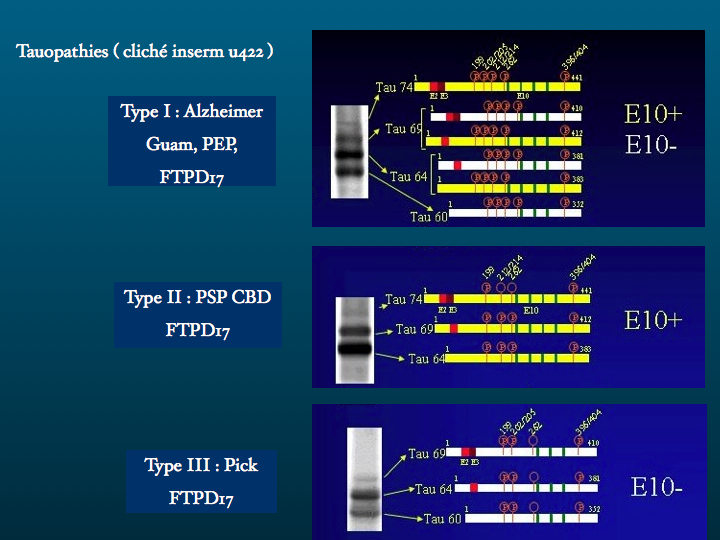
C: dans la maladie dAlzheimer les lésions (tangles, neuropile threads et dystrophic neurites of neuritic plaques) sont composées des six isoformes groupées en PHF (paired helical filaments)
D : dans la FTDP-17, de nombreuses mutations pathogènes provoquent laggrégation des trois isoformes Tau avec 4 répétitions, Tau 4R, qui forment des filaments retrouvés dans les neurones et les astrocytes.
5. Les isoformes tau avec la région "exon 10" se fixent plus fortement à la tubuline, et stabilisent davantage les microtubules.
6. L'interaction tau - tubuline est également régulée par le stade de phosphorylation des protéines tau
 Je vous propose maintenant de
Je vous propose maintenant de




 Revenons au phénotype clinique et à ses liens avec les autres niveaux :
Revenons au phénotype clinique et à ses liens avec les autres niveaux :

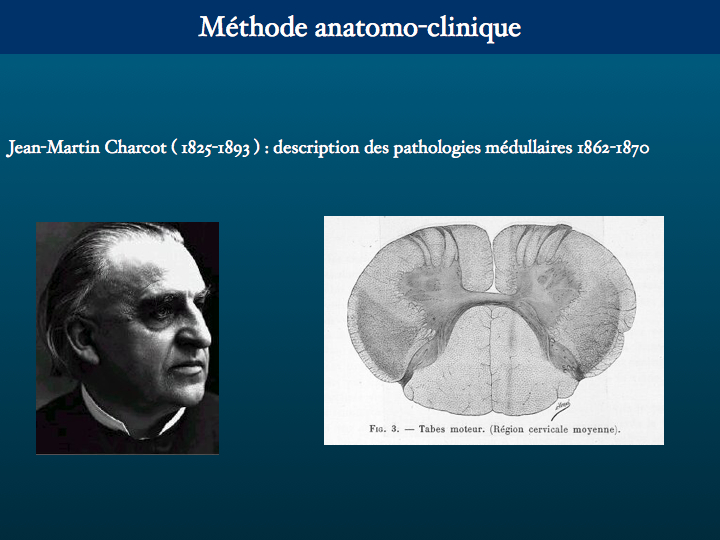
Nous rappelons en passant que toute recherche de nos confrères biologistes moléculaires, généticiens, anatomopathologistes ne peut exister que si un clinicien a amorcé le mouvement en échafaudant une hypothèse diagnostique. Appartenant moi-même à cette dernière espèce, j'ai voulu illustrer la limite de nos possibilités d'interprétation à travers cette oeuvre de Grünewald, réalisée au début du XVIème siècle pour décorer un couvent de moines thérapeutes près de Colmar, en Alsace, et instruire les visiteurs et les malades qu'ils abritaient sur la route des grands pélerinages. Les Antonins soignaient les affections de la peau, entre autres, et notamment le feu de Saint Antoine que l'on sait actuellement provoqué par l'ergot de seigle, avec d'autres manifestations neurologiques, comme des hallucinations, dont souffrait précisément Antoine. Mais pour Charcot, qui s'est intéressé à la Tentation de Grünewald, les lésions cutanées représentées par ce dernier sont en fait typiques de la syphilis.

Le pauvre Antoine, auquel je m'identifie, est soumis à un rude traitement de la part d'une horde de monstres, parmi lesquels je reconnais quelques amis histologistes, biologistes moléculaires, généticiens, et neuro-radiologues bien entendu. Plus sérieusement, les tentations du clinicien sont d'identifier, derrière le phénotype clinique auquel il a seulement accès, le génotype - devant une chorée : c'est une maladie de Huntington, une amyotrophie neurogène familiale : c'est un Charcot-Marie et Tooth ... ; voire le phénotype protéique, le protéotype, ce qui s'avère encore plus délicat.
Mathias Grünewald (1475/1480-1528) Retable d'Issenheim : La Tentation de saint Antoine

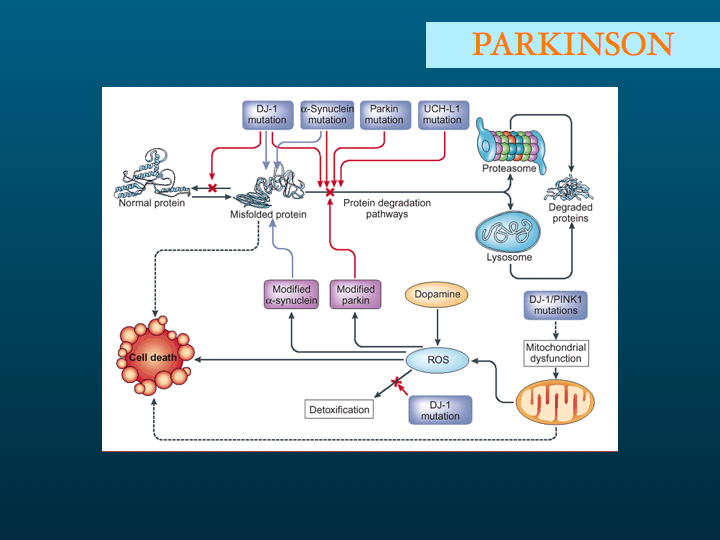
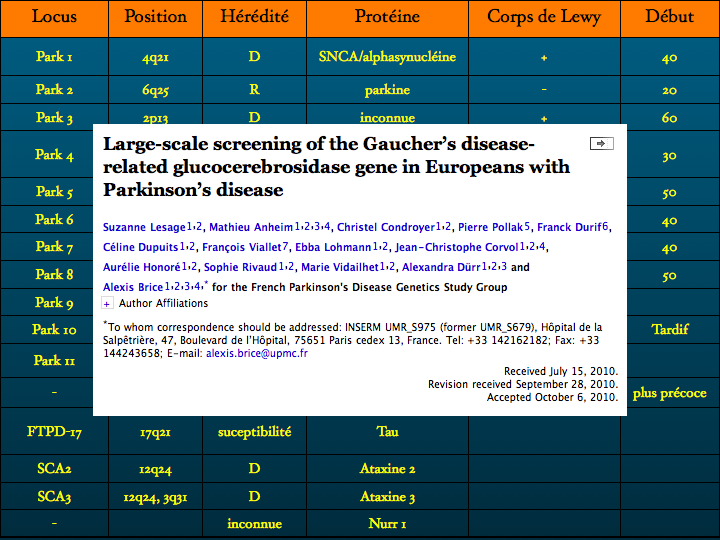
Voyons pour commencer le lien entre le phénotype clinique et le génotype : on peut opposer les formes familiales et les formes sporadiques selon les critères suivants : en faveur des premières, la rareté, un début plus précoce, une progression rapide, et bien évidemment une notion d'affection neurologique familiale ... Rien de très sensible ni de très spécifique. On opposera sur le plan physiopathologique, la production excessive de la protéine pathologique dans les formes familiales, et l'accumulation par défaut de dégradation physiologique dans les formes sporadiques.
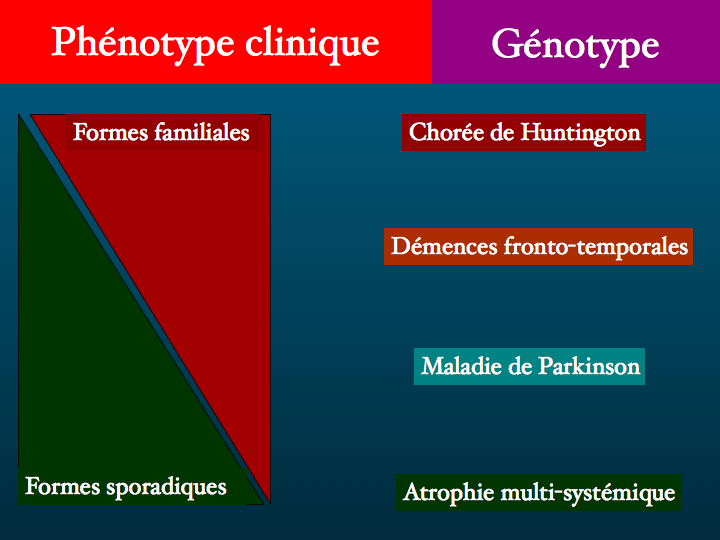
Certaines maladies sont toujours familiales - mais l'anamnèse peut faire défaut : ainsi la chorée de Huntington, ou l'ataxie de Friedreich ; d'autres ne le sont à notre connaissance jamais : aucune forme familiale d'atrophie multi-systémique n'a été publiée. Prenons le groupe cliniquement très hétérogènes des démences frontotemporales que nous étudierons plus en détail dans une semaine : trente à quarante pour cents des observations sont des formes familiales ; en revanche, dans la maladie d'Alzheimer ou la maladie de Parkinson, les formes familiales sont plus rares, environ dix pour cent pour la seconde, un peu moins pour la première. Les formes familiales de paralysie supra-nucléaire progressive et de dégénérescence cortico-basale sont très rares.
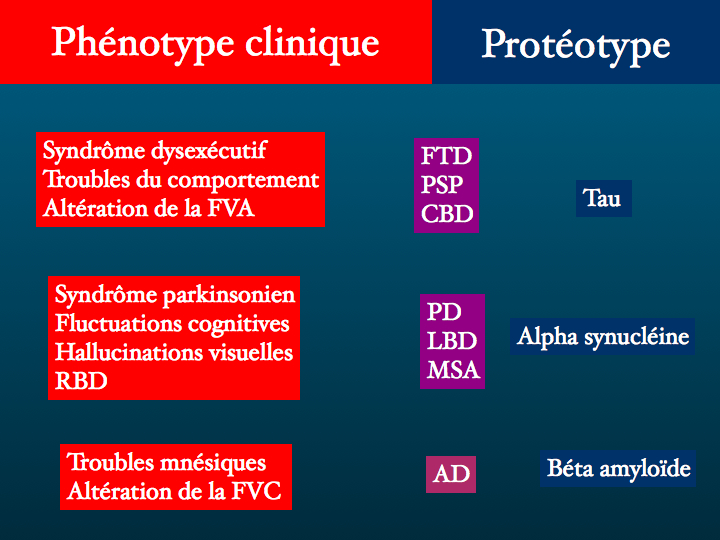
Quant à la tentation qui consiste à inférer à partir d'une présentation clinique le protéotype de la maladie dégénérative en cause, c'est un exercice périlleux, nous allons le vérifier.
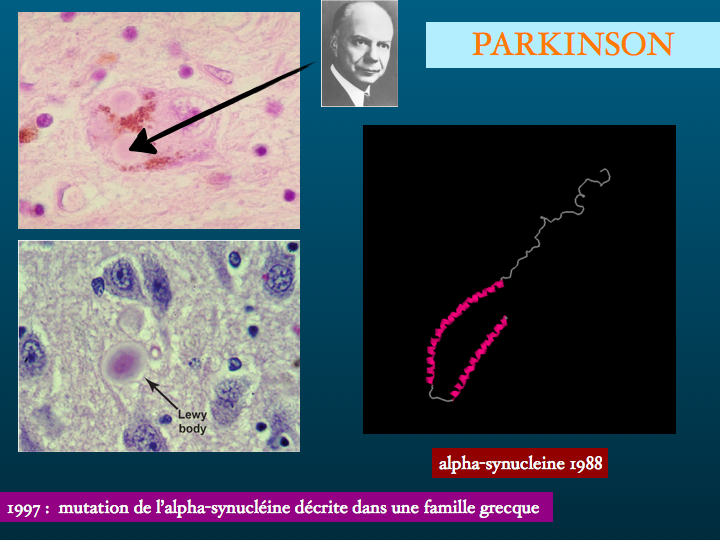
Certes, un syndrôme parkinsonien, des fluctuations cognitives, des hallucinations visuelles, la notion de troubles du sommeil REM, orientent dans l'ordre vers une maladie de Parkinson, une démence à corps de Lewy, une atrophie multisystémique toutes trois étant des alpha synucléopathies. Des troubles mnésiques non améliorés par l'indiçage, une altération de la fluence verbale catégorielle vous orientent vers le diagnostic de maladie d'Alzheimer - pathologie de la beta-amyloïde. Un syndrôme dysexécutif, des troubles comportementaux, une altération de la fluence verbale alphabétique évoqueront une tauopathie.


Normalement, une conférence est faite pour éclairer et non pour obscurcir lintelligibilité du sujet : et si vous éprouvez, au terme de cette introduction, un sentiment de découragement , j'ai manqué mon but, qui est certes de vous placer devant la complexité croissante de nos systèmes de classification, mais aussi de porter à votre connaissance l'existence de clés permettant d'accéder à la compréhension de ces nouveaux systèmes. Il faut accepter l'idée de devenir polyglotte - afin que l'édifice à la construction duquel nous participons tous ici ne s'écroule pas comme la tour de Babel où soudain les hommes, punis pour leur prétention, ne parlèrent plus la même langue et ne purent collaborer plus avant dans leur projet.
À propos de taxonomie et de complexité, j'aime bien rappeler qu'Aristote dans son histoire des animaux traite de moins de cinq cents espèces ; que Buffon, zoologue, en affronta plus de dix mille, et que son contemporain et rival Linné, qui était botaniste, en classa environ 67000 ; qu'actuellement nous évaluons avec beaucoup d'incertitude le nombre d'espèces animales à environ 1,3 millions. Mais le domaine qui de mon point de vue à le plus d'affinité avec celui des neurosciences est l'astronomie : on y travaille dans l'infiniment nombreux, l'inaccessible, l'observation indirecte, et les galaxies sont seulement trois fois plus nombreuses que les neurones d'un homo sapiens standard.

Si un jour vous passez par Nice, où je vis, pensez à vous promener une nuit du côté de lObservatoire inauguré en 1887, conçu par Garnier, et dont la coupole est luvre de Gustave Effel. Les astronomes sont accueillants, et nous apprennent à déchiffrer leur ciel.

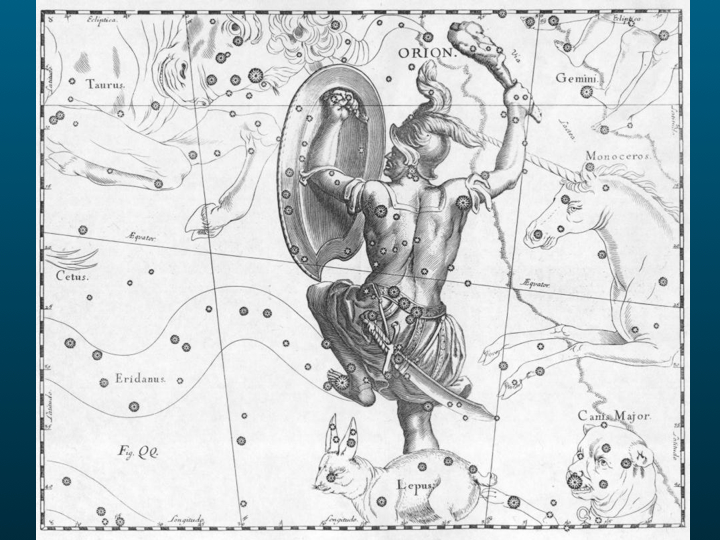
En hiver, vous verrez en le contemplant un groupe détoiles dont la forme fixe a très tôt attiré le regard : Bételgeuse, Rigel, sont des noms qui vous sont sans doute familiers même si vous ignorez letymologie : ils sont formés à partir des expressions arabes signifiant lépaule et la jambe gauche du Géant ; dans lantiquité grecque les premiers observateurs du cosmos lui donnèrent le nom de constellation dOrion, un chasseur qui fut tué par un scorpion sur lordre dHéra.

Maintenant ce n'est plus une constellation figurant le chasseur maudit par Héra, et qui pour consolation est placé parmi les astres à l'opposé du Scorpion qui ne l'atteindra plus jamais, mais une nébuleuse, un fantastique nuage moléculaire. Les lois qui régissent les interactions de ces molécules sont d'une complexité incomparable aux principes qui gouvernaient les simples mouvements des planètes.
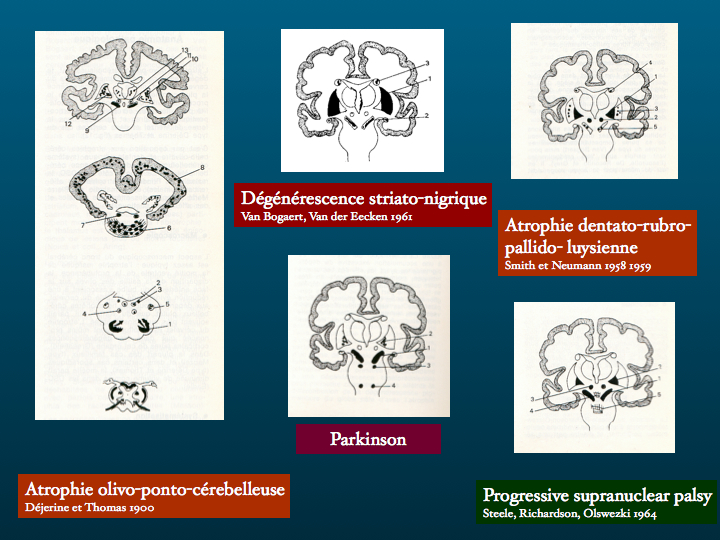
La confortable constellation des syndrômes parkinsoniens, nous la parcourions de proche en proche, guidés par le traité de Richard Khalil, en nous berçant pour certains de l'illusion d'un continuum que nous découvrions peu à peu, par bribes, au fur et à mesure de l'identification des variétés de pathologies neuro-dégénératives.
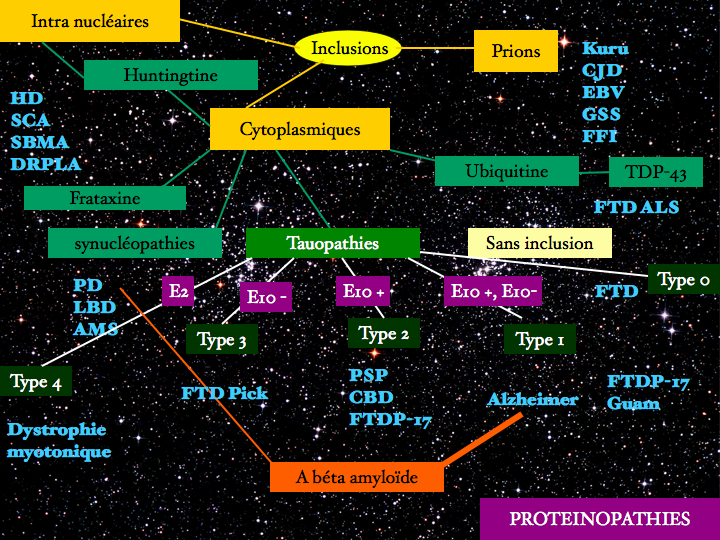
Au catalogue, à linventaire, de variétés cliniques apparemment solidement définies, à cette constellation que je vous décrivais en début dexposé, les généticiens, les anatomopathologistes et les biologistes moléculaires ont substitué une nébuleuse autrement plus complexe nous obligeant à repenser fondamentalement les relations de causalités entre divers ordres de phénomènes. Certains concepts ont pour fonction de nommer à des fins taxinomiques les productions mentales : les maladies dégénératives existent-elles, ou sont elles comme Orion le chasseur une vue de lesprit ? On se tiendra ici à la position nominaliste dAristote, celle adoptée par Buffon classant les animaux : la notion despèce nexiste pas en soi, pas plus que celle de maladie neuro-dégénérative. Mais alors à quoi bon se payer de mots, conserver une expression dont le sens semble se résumer au catalogue des affections rangées sous son enseigne ?

Au catalogue des maladies neurodégénératives, dont la classification fondée sur des principes de ressemblances et de différences comme s'est substitué une taxonomie fondée sur un mécanisme moléculaire commun : le regroupement de séquences protéiques ayant une conformation anormale (misfolding) riche en feuillets plissés béta formant des fibrilles.
Nous allons maintenant aborder la question des nouveautés conceptuelles dans une seconde partie intitulée : from degeneration to degeneracy.